Paralisi spastica ascendente ereditaria a esordio infantile
- 1 Progetti di Ricerca Finanziati
- 1 Ricercatore
- 49.875€ Finanziamento totale
Che cos’è e come si manifesta la paralisi spastica ascendente ereditaria a esordio infantile?
- La paralisi spastica ascendente ereditaria a esordio infantile (IAHSP) è una malattia molto rara che colpisce i motoneuroni, le cellule nervose che impartiscono ai muscoli il comando di movimento. Alla nascita questi pazienti sono normali, così come nelle prime fasi dello sviluppo. Tuttavia, già nei primi due anni di vita compaiono paraplegia spastica, debolezza e rigidità degli arti inferiori, che entro i 7-8 anni si estendono anche agli arti superiori. Si sviluppano inoltre difficoltà nell’articolare le parole (disartria) e nell’alimentarsi (disfagia, a volte associata anche allo sbavamento), oltre che lentezza nei movimenti oculari. La maggior parte dei pazienti diventa dipendente dalla sedia a rotelle prima della tarda infanzia o della prima adolescenza e alcuni presentano difficoltà alimentari (deglutizione dei liquidi) che esordiscono nella seconda decade. Successivamente, la malattia progredisce verso la tetraplegia spastica grave e si può manifestare una sindrome pseudo-bulbare. In genere sono preservate le funzioni cognitive. La prognosi è buona e la sopravvivenza nella maggior parte dei casi è a lungo termine, tuttavia la qualità della vita è compromessa dai segni neurologici progressivi e dalla perdita dell'indipendenza. Finora sono state descritte 17 famiglie comprendenti una trentina di casi.
Come si trasmette la paralisi spastica ascendente ereditaria a esordio infantile?
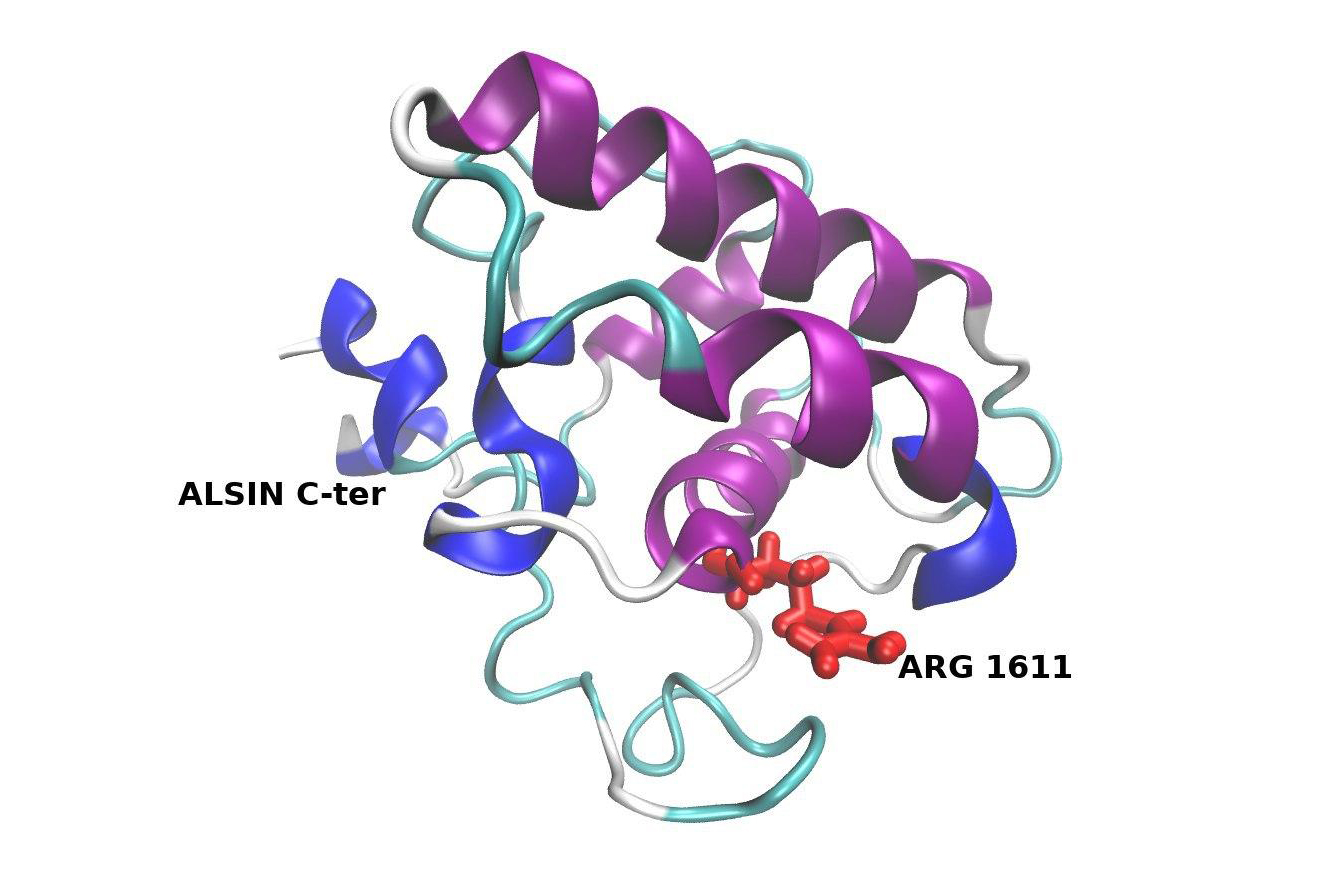
- La IAHSP è causata da mutazioni del gene ALS2, che codifica per l’alsina, una proteina molto presente nei motoneuroni la cui funzione non è ancora del tutto nota. Si trasmette con modalità autosomica recessiva: per manifestare i sintomi bisogna cioè ereditare il difetto da ciascuno dei genitori, entrambi portatori sani. Una coppia di portatori ha una probabilità del 25 per cento di avere figli malati. Tuttavia, sebbene questo sia l’unico gene associato alla malattia finora identificato, sono stati descritti casi di pazienti affetti che non presentano mutazioni in ALS2.
Come avviene la diagnosi della paralisi spastica ascendente ereditaria a esordio infantile?
- La diagnosi si basa sui segni caratteristici della malattia e sulla progressione ascendente. Gli studi elettrofisiologici mostrano una disfunzione grave dei potenziali evocati motori, mentre risultano normali i potenziali evocati somato-sensoriali, l'elettromiografia e le velocità di conduzione nervosa. La risonanza magnetica è normale nei bambini affetti, ma può mostrare alterazioni cerebrali in età avanzata. La diagnosi può essere confermata dall’analisi molecolare. La diagnosi differenziale si pone con altre malattie dei motoneuroni come la sclerosi laterale primitiva giovanile e la sclerosi laterale amiotrofica giovanile. È inoltre possibile la diagnosi prenatale nelle gravidanze a rischio, se sono note le mutazioni nella famiglia.
Quali sono le possibilità di cura attualmente disponibili per la paralisi spastica ascendente ereditaria a esordio infantile?
- Attualmente non è disponibile un trattamento specifico e la presa in carico consiste principalmente nella fisioterapia e nella terapia occupazionale (o ergoterapia) per favorire la mobilità e l'indipendenza.
Ultimo aggiornamento
02.09.20
Approfondisci su Orphanet
18.12.20
Famiglie in prima linea contro le malattie genetiche rare, al fianco della ricerca
Per i pazienti con malattie genetiche rare e le loro famiglie spesso la ricerca rappresenta l’unica speranza. Ma le stesse famiglie possono contribuire in modo importante al lavoro dei ricercatori.

14.09.20
Seed Grant: a un bioingegnere i fondi messi a disposizione dell’associazione Help Olly
Marco Agostino Deriu del Politecnico di Torino costruirà al computer un modello rappresentativo della struttura della proteina che causa la paralisi spastica ascendente ereditaria ad esordio infantile per indirizzare in modo più mirato la ricerca su potenziali strategie terapeutiche.
09.09.20
Da associazioni pazienti e Fondazione Telethon 350 mila euro per 7 progetti di ricerca sulle malattie genetiche rare
Finanziati gruppi di ricerca delle Università di Napoli, Bologna, Verona e Torino, dell’Istituto Neurologico Carlo Besta e degli Istituti Telethon Tigem di Pozzuoli e SR-Tiget di Milano
